All published articles of this journal are available on ScienceDirect.

Isoflurane but not Fentanyl Causes Apoptosis in Immature Primary Neuronal Cells
Authors Info & Affiliations
Abstract
Background:
Anaesthetics are widely used in new-borns and preterm infants, although it is known that they may adversely affect the developing brain.
Objective:
We assessed the impact of the volatile anaesthetic, isoflurane, and the intravenous analgesic, fentanyl, on immature and mature embryonic neuronal cells.
Methods:
Primary neuronal cultures from embryonic rats (E18) cultured for 5 (immature) or 15 days (mature) in vitro (DIV), respectively, were exposed to isoflurane (1.5 Vol.%) or fentanyl (0.8 - 200 ng/ml) for 24 hours. Experiments were repeated in the presence of the γ-amino butyric acid-A (GABAA) receptor antagonists, bicuculline or picrotoxin (0.1 mmol/l), or the pancaspase inhibitor zVAD-fmk (20 nmol/l). Cell viability was assessed by methyltetrazolium (MTT) metabolism or lactate dehydrogenase (LDH) release.
Results:
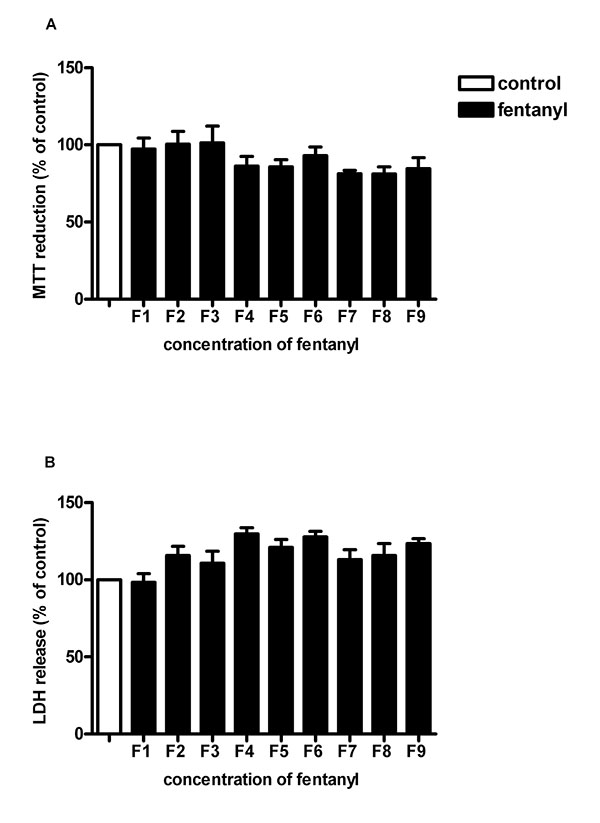
Isoflurane reduced cell viability significantly in primary neuronal cells cultured for 5 DIV (Δ MTT -28 ±13%, Δ LDH +143 ±15%). Incubation with bicuculline, picrotoxin or zVAD-fmk protected the cells mostly from isoflurane toxicity. After 15 DIV, cell viability was not reduced by isoflurane. Viability of primary neurons cultured for 5 DIV did not change with fentanyl over the wide range of concentrations tested.
Conclusion:
Immature primary neurons may undergo apoptosis following exposure to isoflurane but are unaffected by fentanyl. Mature primary neurons were not affected by isoflurane exposure.
1. INTRODUCTION
There is controversial epidemiologic evidence to suggest that children who were exposed to anaesthesia as neonates or infants carry an elevated risk of adverse long-term behavioral or neurodevelopmental outcomes [1-6].
Intravenous anaesthetics that block N-methyl-D-aspartate (NMDA) glutamate receptors, such as ketamine, or are γ-amino butyric acid-A (GABAA) receptor agonists, e.g. benzodiazepines, are known to cause apoptotic death of neurons in the brains of newborn animals [7, 8], associated with long-term cognitive deficits [9]. Induction of general anaesthesia in infants mostly involves volatile anaesthetics. In newborn rodents, pigs or non-human primates, an increase of neuronal cell death has been observed following isoflurane exposure [10-17], with rather subtle long-term cognitive dysfunction occurring in a partially sex-disparate fashion [4, 18, 19]. Isoflurane-induced neuronal apoptosis appears to occur only at a certain stage of neuronal development, which is dependent on the age of the neuron but independent of the age of the animal [15]. Isoflurane, however, appears to have protective properties as well. Survival of cortical neurons subjected to transient oxygen-glucose deprivation has been found to improve with isoflurane post-conditioning [20]. Moreover, there appears to be reduced brain damage in mice exposed to isoflurane before or after ischemic injury [21, 22] or in rats who received isoflurane after induction of germinal matrix hemorrhage [23].
Exposure to isoflurane, which affects various cell types outside and inside the brain [8, 24], causes a panoply of cardiorespiratory and metabolic side effects [10]. Its use in newborn rodents is associated with considerable mortality [25]. Therefore, we aimed to characterize the immediate action of isoflurane on isolated immature and mature neurons.
2. METHODS
All experiments were carried out in accordance with ethical principles and guidelines for experiments on animals and were approved by the animal welfare committees of the Berlin State Office for Health and Social Affairs (LaGeSo), Germany (Reg 0163/03).
Primary neuronal cells were prepared from the cortices of Wistar rat embryos at day 18 (E18) (Forschungseinrichtung für Experimentelle Medizin, Charité - Universitätsmedizin Berlin).
2.1. Cell Preparation
The cerebral cortices were delivered from the meninges and cells were dissociated by trypsin-EDTA in phosphate buffered saline (PBS) for 15 minutes at 37°C. Cells were subsequently resuspended in serum-free neurobasal medium supplemented with 2% B27-supplement (Gibco, Invitrogen, Karlsruhe, Germany), 1% L-glutamine (Sigma-Aldrich, Taufkirchen, Germany) and 1% penicillin/streptomycin (Biochrom, Berlin, Germany). We homogenised the tissue with fire-polished Pasteur pipettes by aspirating several times followed by a centrifugation (1200 U/min) for 2 minutes at room temperature. The resulting pellet was resuspended in medium at 1 x 106 /ml and seeded at 96-well-plates coated with poly-D-lysine (0.5% w/v in water; Sigma-Aldrich) 100 µl/ well. Cells were incubated at 37°C in a humidified atmosphere (5% CO2, 95% air). Medium was half-changed every 6th day.
2.2. Isoflurane
After either 5 DIV (immature cells) or 15 DIV (mature cells), the plates were placed in an incubator chamber (Billups-Rothenberg, Del Mar, California, USA) and flooded with 1.5 Vol.% isoflurane (Forene®, Abbott, Wiesbaden, Germany) through a vaporizer (Vapomat 6; STEPHAN Medizintechnik, Gackenbach, Germany) in a gas of 5% CO2 and 95% air. The gas concentration in the chamber was analysed continuously by a Capnomac gas monitor (Datex-Ohmeda, Helsinki, Finland). By reaching a steady state of 1.5Vol% isoflurane with a flow rate of 6 l/min the saturated chamber was sealed airtight and placed in the same incubator as control cell the for 24 hours.
2.3. Fentanyl
Neuronal cells for 5 DIV were exposed to fentanyl (Fentanyl® Janssen; Janssen Cilag, Neuss, Germany) in various concentrations from 0.8 to 200 ng/ml for 24 hours. We prepared a stock solution of 400 ng/ml in medium and made a serial dilution in medium (0.8 ng/ml; 1.56 ng/ml; 3.125 ng/ml; 6.25 ng/ml; 12.5 ng/ml; 25 ng/ml; 50 ng/ml; 100 ng/ml; 200 ng/ml). This cell culture model allowed us to study the effect of high dosages of fentanyl, which are difficult to assess in non-ventilated newborn rodents due to the respiratory depression caused by fentanyl.
2.4. Inhibition
To examine the role of GABAA receptors, cells were incubated with the GABAA receptor antagonists bicuculline (0.1 mmol/l; Sigma-Aldrich) and picrotoxin (0.1 mmol/l; Sigma-Aldrich) 30 min prior to isoflurane exposure.
To characterize the type of cell death, cells were treated with the pan-caspase-inhibitor zVAD-fmk (benzyloxycarbonyl-valin-alanin-aspartate-fluoromethylketon; Sigma-Aldrich) prior to exposure to isoflurane. zVAD-fmk inhibits all caspases that are participating in apoptosis but does not affect necrosis.
2.5. MTT Assay
In the methyltetrazolium (MTT) assay the mitochondrial reductase only present in metabolically active cells reduces the yellow coloured 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to purple formazan. 10 µl MTT (Sigma-Aldrich) at a final concentration of 0.5 mg/ml was added to each well containing 1x106 cells as described before. After 2 hours the insoluble formazan was dissolved into a coloured solution by adding 100 µl of 10% sodium dodecyl sulphate. Absorbance was measured by a microplate reader (Bio-Rad, München, Germany) at 570 nm with a reference wavelength of 630 nm. The amount of MTT formazan is directly proportional to the number of living cells [26].
2.6. LDH Release Assay
The lactate dehydrogenase (LDH) release assay (Cell Death Detection ELISAPLUS; Roche, Grenzach-Wyhlen, Germany) was used to assess the amount of LDH enzymes released into the medium by cells undergoing lysis. Therefore, the cell-free supernatant was removed from exposed plates and mixed with a catalyst (diaphorase) and a dye solution (iodonitrotetrazolium chloride (INT) and sodium lactate) from the cytotoxicity detection kit. By the oxidation of lactate to pyruvate the released LDH in the supernatant reduces NAD+ to NADH+H+. The added catalyst transforms INT to the coloured formazan by the oxidation of NADH+H+ to NAD+. At a wavelength of 495 nm with reference to 630 nm the absorbance was quantified with a microplate reader. The amount of formazan product is directly proportional to the enzyme activity [27].
2.7. Statistical Analysis
GraphPad Prism 4.03 software (GraphPad Software, La Jolla, CA, USA) was used for all tests. Results were compared with the one-way ANOVA test followed by Bonferroni`s multiple comparison test at a level of significance of P<0.05. Experiments were repeated as indicated. Data are presented as mean ± standard error of the mean (SEM).
3. RESULTS
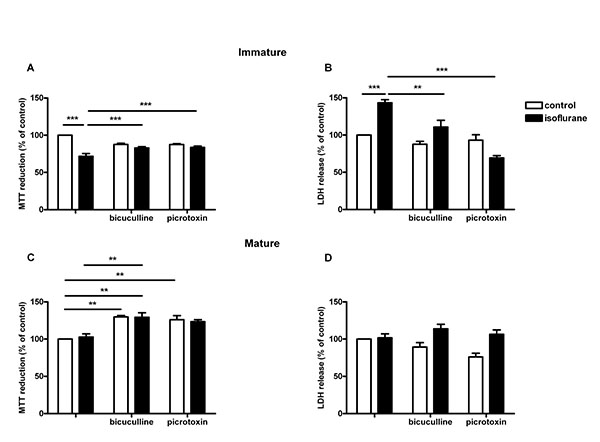
3.1. Immature Cells Exposed to Isoflurane
As a marker of cell viability the MTT assay (Fig. 1A) showed a significant decrease of cell viability of the cultures exposed to 1.5 Vol.% isoflurane in comparison to the untreated control group (71.7 ± 3.8%; P<0.001). The addition of the GABAA-receptor antagonists lead to a significant increase in cells exposed to isoflurane in combination to the antagonists (bicuculline 83.0 ± 1.6% and picrotoxin 83.8 ± 1.7%; P<0.001).
The LDH release assay (Fig. 1B) showed a significant elevation of LDH release of the immature neurones exposed to isoflurane (143.3 ± 4.3%, P < 0.001) in comparison to the untreated control cells. Cells treated with isoflurane incubated with GABAA-receptor antagonists showed a significant reduction of cell death (bicuculline 118.2 ± 6.4%, P<0.01 and picrotoxin 73.1 ± 3.3; P<0.001). There were no significant differences between control cells and untreated cell incubated with bicuculline or picrotoxin.
3.2. Mature Cells Exposed to Isoflurane
There was no significant reduction in cell viability of mature cells (15 DIV) that were exposed to isoflurane (105.4 ± 3.7, ns) (Fig. 1C). Cell viability increased in control cells incubated with bicuculline (129.8 ± 1.9%; P<0.01) and picrotoxin (126.2 ± 5.3; P<0.01) as well as in cells exposed to isoflurane and bicuculline (129.3 ± 6.0%; P<0.01) compared to isoflurane treated cells. For cells treated with isoflurane and picrotoxin the increase was not significant (122.1 ± 3.0).
Mature cells exposed to isoflurane showed no significant increase in cell death, measured by LDH release compared to controls (101.8 ± 4.5) (Fig. 1D). Control cells incubated with bicuculline or picrotoxin showed no significant decrease to untreated cells nor did isoflurane treated cells in presence of GABAA-receptor antagonists.
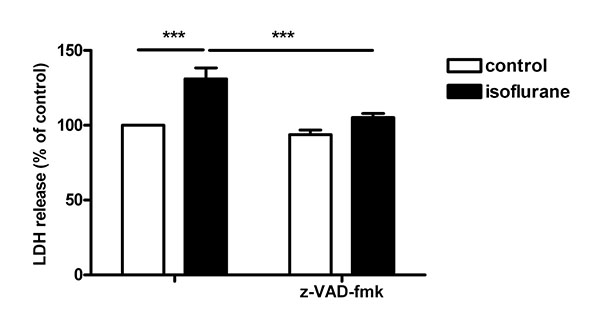
3.3. Inhibition of Apoptosis in Immature Cells
Exposure of immature neurons (5 DIV) showed increased LDH released compared to controls. The addition of the pan-caspase-inhibitor zVAD-fmk (Fig. 2) to the cultures neutralized the adverse effect of isoflurane on cell viability (135.3 ± 3.2 vs. 109.1 ± 2.1%, P<0.001).
CONCLUSION
The results of the present study indicate that isoflurane damages a proportion of freshly isolated immature neurons of embryonic rats cultured for 5 days but not mature neurons cultured for 15 days. Isoflurane-induced cell death is inhibited by the pan-caspase inhibitor, zVAD-fmk, pointing to apoptosis as its principal mode. The toxic action of isoflurane can be partially inhibited by bicuculline or picrotoxin, suggesting involvement of the GABAA receptor pathway. Viability of immature neurons is not affected by increasing doses of fentanyl.
The results obtained with isolated neuronal cells are in line with what is known from experiments involving newborn animals being exposed to isoflurane. In newborn mice, general anaesthesia with 1.5 Vol.% isoflurane has been found to cause a 4-11 fold increase of neuronal death in various brain regions, as compared to controls.[10, 28, 29] A 6-hour isoflurane exposure eliminates approximately 2% of cortical neurons [14]. Apoptosis appears to be the predominant mode of isoflurane-induced death, as evidenced by caspase-3 cleavage [10]. Notably, mice genetically deficient in the apoptosis-triggering cell surface receptors Fas (CD95) or FasL (CD95L) show reduced isoflurane-induced neuronal cell death [30]. Isoflurane susceptibility vanishes in juvenile and adult animals except for brain areas displaying continued neurogenesis [28].
The neuronal apoptosis following neonatal isoflurane exposure is linked to altered behaviour and memory functions, with greater long-term cognitive impairment after multiple neonatal exposures to isoflurane than after a single exposure [18]. In addition to directly driving neurons into apoptotic cell death, isoflurane also impairs the capacity of astrocytes to support neuronal development [31] and evokes errors in axon targeting and growth cone guidance in the developing neocortex in a GABAA receptor-mediated fashion [32]. Our experiments using bicuculline, a competitive GABAA receptor antagonist, and picrotoxin, a non-competitive GABAA receptor antagonist, demonstrate the involvement of the GABAA receptor pathway in mediating the apoptosis-inducing action of isoflurane. The differing effects of bicuculline and picrotoxin might be explained by the modulation of the GABAA receptor by isoflurane by increasing the affinity and that there seems to by a blocking action at or near the picrotoxin site [33].
Experimentally, the toxic effect of isoflurane for the developing brain extends to non-human primates. Brains of both fetal [24] and neonatal rhesus macaques [8, 11] display signs of neuronal apoptosis upon exposure to isoflurane (0.7 -1.5 Vol.% for 5 hours). In humans, there is epidemiological evidence to suggest a modestly elevated risk of adverse behavioural or developmental outcomes in children who were exposed to anaesthesia during early childhood [1, 3, 4]. As all retrospective observational approaches, these cohort database analyses are tainted with confounding variables [34], but randomized controlled trials are about to yield answers [35]. Neurodevelopmental changes induced by anaesthetics are subtle and therefore require careful controls to be proven. Rather than causing gross or fine motor impairments which can be diagnosed by standardized examinations at 2 years of age, exposure to anaesthetics at very early age appears to preferentially provoke long-term deficits in cognition, language and executive functions [2, 5] which cannot be reliably assessed prior to school age.
Various aspects emerging from the epidemiological data correspond to the results obtained in the experiments with isolated immature neuronal cells. Susceptibility of the cells to isoflurane was seen only in immature cells, while cells cultured for 2 weeks became resistant to the action of isoflurane. There seems to be a critical period of cellular development during which neurons are susceptible to anaesthesia-induced apoptosis. Vulnerability towards anaesthetics reflects the age of the neurons rather than the age of the organism [15] and is linked to ongoing neurogenesis [28]. In human infants, susceptibility to anaesthetics appears to be restricted to early ages, with very preterm infants exposed before term being at greatest risk [36, 37]. Children exposed to surgery and general anaesthesia prior to 3 years of age had somewhat lower scores than their unexposed peers in receptive and expressive language, even with a single exposure to anaesthesia [2], as did healthy school-aged children and adolescents who had undergone surgery with anaesthesia before 4 years of age [5]. In contrast, exposure to anaesthesia at a later age had no measurable effects on language or cognitive function [38].
Brief exposure times (< 1h) to sevoflurane in vivo had shown to carry little or no risk in a randomized controlled trial involving human infants with a postmenstrual age < 60 weeks undergoing herniorrhaphy [35]. However, in term infants undergoing complex cardiac surgery, a positive association between volatile anaesthetic exposure (isoflurane), MRI-assessed brain injury, and lower neurodevelopmental outcome scores at 12 months of age were demonstrated [39]. In term infants undergoing major non-cardiac surgery, cognitive and motor developmental delay at 2 years of age was found in less than a quarter of the children assessed, with neurodevelopmental outcome scores on average 0.5 SD below the normative score of the healthy population [6].
We also studied fentanyl over a wide range of concentrations, initiated by conflicting reports about the effects of fentanyl in brains of adult rats [40-42]. Freshly isolated human lymphocytes may undergo apoptosis when incubated with fentanyl [43] but fentanyl did not affect cell viability of immature neuronal cells 5 DIV. We used concentrations from 0.8 to 200 ng/ml of fentanyl in cell cultures that correspond to plasma concentration levels of high-dose fentanyl application in adults [44-46] and neonates [47]. Our results are in line with experiments involving mechanically ventilated newborn piglets which showed no signs of neuronal apoptosis after receiving fentanyl administered at a continuous intravenous infusion over 24 hours at dosages of 50-75 µg/kg/h. In contrast, control animals receiving 2 Vol.% isoflurane showed widespread neuronal apoptosis [13]. Animal experiments using moderate doses of fentanyl in newborn rats support the neonatal use of fentanyl even in premature newborns [48]. In very low birth weight human infants, no association between cumulative fentanyl dosage and neurodevelopmental outcomes has been found after controlling for other variables [49].
In summary, the data presented here add weight to considerations to exert caution when using volatile anaesthetics in very preterm infants. Maturation of brain neuronal cells appears to confer resistance to the apoptosis-promoting action of isoflurane but establishing an age limit for safe use of volatile anaesthetics is awaiting further clinical investigations. The apparent lack of neurotoxicity of fentanyl encourages attempts to prioritize opioids over GABAA agonists when designing anaesthesia protocols for preterm infants.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
All animal experiments were performed in accordance with international guidelines for good laboratory practice and were approved by the animal welfare committees of Berlin, Germany. (Reg 0163/03).
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

